Laboratory of Macromolecular Crystallography
This is a review of the works carried out in LMC of the IMPB
RAS. Information on other papers in this field may be found in the original
papers listed below.
New protocol for the package AMoRe
(T.Petrova, 2003)
The main idea of using the method of Molecular Replacement in the crystallography of macromolecules
You can find a few good crystallographic courses with detailed explanations of
Molecular Replacement technique ( see, for example,
http://www-structmed.cimr.cam.ac.uk/Course/MolRep/molrep.html ). The main idea
is to build a tentative crystal structure using known molecular model with the
high level of sequence identity with the unknown structure. This model is
oriented and possessed in the unit cell of unknown structure in order to start
model building and refinement.
Two ways to work with the package AMoRe
There are two ways to work with the package AMoRe (J.Navaza): - to run manually
AMoRe procedures one after another; the values of input parameters are edited by
user depending on the results of the previous steps; - to use an automated
protocol, which runs sequentially all procedures for the particular MR problem.
The first way is universal. However, it requires the knowledge of the MR method
and is time consuming. Therefore, using the automated protocol is more popular.
The original automated protocol of AMoRe (called 'job') is simple and finds a
correct solution in the vast majority of MR cases. However, some MR problems
could be solved only by the first way. The new automated protocol was developed
mostly for these difficult cases. The new protocol is also supposed to take
optimal time; i. e., it stops searching as soon as an acceptable solution is
found and performs an exhaustive search in difficult cases. Our preliminary
tests have shown that the new protocol is able to solve automatically several
difficult cases that would otherwise require laborious manual processing.
However, this protocol is not universal, and some cases cannot be solved
automatically.
Main features
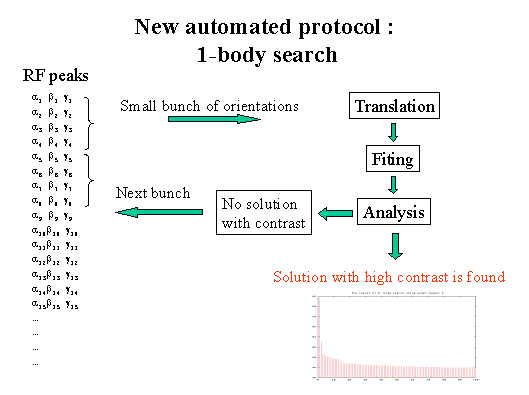
- After each running of procedures TRAING and FITING, an analysis of the
number of solutions obtained is performed.
- The criteria of a correct solution are the highest value of the correlation
coefficient and a high value of the contrast.
- To optimise the total time of the search, RF output orientations are
included into the next search by big bunches. These big bunches are subdivided
into small bunches by which the orientations for the translation search are
selected. The orientations of each next bunch are selected only if the solution
has not yet been found.
- In the n-body case, different models can be placed into the unit cell either
in parallel or sequentially.
- In the case of possible translational NCS-symmetry, molecules can be
translated by pairs.

Software requirements
- AMoRE: last version ( as of July 2002 ) is available through anonymous FTP
from:
ftp://amaril.gv.cnrs-gif.fr/pub/jorge
- awk (GNU awk recommended)
- Gnuplot (for graphics)
- Rasmol (used to show the final solution)
Installing
The protocol is distributed as a compressed file auto_amore-1.0.tar.gz . It is
available through anonymous FTP
from:
ftp://amaril.gv.cnrs-gif.fr/pub/auto_amore
The following commands will uncompress and unpack the archive.
On machines running Linux, just type:
tar xvzf auto_amore-1.0.tar.gz
otherwise (SGI, DEC, SunOS, etc...):
gunzip auto_amore-1.0.tar.gz
tar -xvf auto_amore-1.0.tar
This will create the directory auto_amore-1.0/ in the current directory.
Running
Before running the protocol, you must install AMoRe and define all necessary
environmental variables as described in the AMoRe documentation (see the file
tutorial.ps).
The automated protocol must be executed from the current working directory. If
the alias auto_amore has been defined as mentioned above, the running is
actuated by the command:
./auto_amore
The protocol operates in the interactive mode; i.e., it requires the user input
for processing either in the form of a question, which must be answered, or a
file which could be edited (optional).
User decisions at run-time
The protocol requires the user decision in the following cases: when a strong
Patterson function peak (peaks) that could indicate the presence of
translational symmetry is (are) found. You can choose either to confirm to use
NCS-symmetry or to use the usual translation. In the first case, for each
orientation, the molecules will be translated by pairs shifted by the known
vector.
When no acceptable solution is found after the 1-body search. You can choose
either to continue searching or to stop the current run in order to change the
input model, the resolution range or to use CC for the 1-body translation. If
this is the first run, it is highly recommended to let the protocol investigate
further, since in many cases after one-body search there is no solution with
strong contrast.
When no final solution could be found using the current big bunch of RF-output
orientations. You are then asked to continue the search using the next big bunch
of orientations or to stop the current run.
Bug reports
Please, report any problem to
auto_amore@mail.ru .
Miscellaneous
This protocol and documentation were written by Tatiana Petrova in the
Laboratoire de Genetique des Virus, CNRS Gif-Sur-Yvette, France, under the
supervision of Jorge Navaza. This work was supported by the European Community
grant in the framework of AUTOSTRUCT project:
http://www.ccp4.ac.uk/autostruct/index.html
March, 24, 2003
|
