Laboratory of Macromolecular Crystallography
This is a review of the works carried out in LMC of the IMPB RAS. Information on other papers in this field
may be found in the original papers listed below.
A statistical approach to the estimation of errors in the phases calculated from
preliminary atomic models.
(1982-1985, 1994-2000)
The goal of this project was to develop an approach, which would allow an
estimation of the errors inherent in the structure factor phases calculated with
the use of a preliminary atomic model of the studied object. By the reasons
discussed below the two parts of this study were separated by about 10 years.
Iterative building of a preliminary atomic model
The traditional goal of the first stage of determining a macromolecular
structure is to find the function ρ(h,k,l), which presents the
electron density distribution in the crystal of a studied object. This function
is periodical in three space directions and may be presented as a
three-dimensional Fourier series
(1)
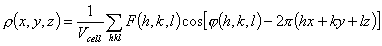
In crystallography, the complex coefficients
F(h,k,l)exp[i φ(h,k,l)] are referred to as
structure factors while real values of F(h,k,l) and
φ(h,k,l) are called magnitudes and phases, respectively. In a
conventional X-ray experiment one can only determine the magnitudes
F(h,k,l). The problem of restoring the phase values is called the
phase problem of X-ray crystallography. Obviously, to solve it, some additional
information on the studied object is required. Once approximate phase values
have been found, they (together with experimental magnitudes) can be used to
calculate an approximate density distribution by formula (1). If only a finite
number of structure factors is used to calculate the series (1) they say that
Fourier synthesis of a finite resolution has been calculated. The synthesis
resolution depend on the number of structure factors used. The more terms in the
series (1) are used the more fine details may be recognised in analysing this
synthesis.
The distribution found is then used to build a preliminary atomic model, i.e. to
find approximate coordinates
Fatal error: Function name must be a string in /home/i/impbran/impb.tmweb.ru/tpl/templates_c/356/3566525821/0.php on line 59
|
