Laboratory of Macromolecular Crystallography
This is a review of the works carried out in LMC of the IMPB RAS. Information on other papers in this field
may be found in the original papers listed below.
Development of the Few Atom Model method for solution of the phase problem
in protein crystallography.
(1994-1998)
This project was developed in collaboration with the Laboratory of Biological
Structures of the Institute of Genetics and Molecular and Cellular Biology
(IGBMC) (Strasbourg, France, PI Prof. A.D.Podjarny). The project was aimed at
generalising the procedure of low-resolution ab-initio phasing, which was
initially developed to manage the information on Fourier syntheses histograms
[1].
The traditional goal of the first stage of determining a macromolecular
structure is to find the function ρ(x,y,z), which presents the
distribution of electron density in the crystal of a studied object. This
function is periodical in the three space directions and may be presented as a
three-dimensional Fourier series
(1)
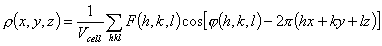
In crystallography, the complex coefficients
F(h,k,l)exp[iφ(h,k,l)] are referred to as
structure factors while real values of F(h,k,l) and
φ(h,k,l) are called magnitudes and phases, respectively. In a
conventional X-ray experiment one can only determine the magnitudes
F(h,k,l). The problem of restoring the phase values is called the
phase problem of X-ray crystallography. Obviously, some additional information
on the studied object must be attracted to solve this problem. Once approximate
phase values have been found, they (together with experimental magnitudes) may
be used to calculate an approximate density distribution by formula (1). If only
a finite number of structure factors are used to calculate the series (1) they
say that the Fourier synthesis of a finite resolution is calculated. The
synthesis resolution depends on the number of structure factors used. The more
terms in the series (1) are used the more fine details may be recognized in
analysing this synthesis. Low-resolution syntheses are calculated with the use
of a small number of structure factors having the smallest indices hkl. Such
syntheses allow one to locate the molecules in the crystal cell and to have the
first information on the molecule shape.
The approach discussed is based on the hypothesis that a low-resolution Fourier
synthesis may be approximated by a small number of "broad" gaussian functions.
These functions may be considered as some huge pseudo-atoms or "blobs". Below we
call such approximations Few Atom Models (FAM). The phases calculated from these
blobs may be used as a reasonable approximation for low-resolution phases. In
favourable cases even a one-blob approximation may provide a dozen of rather
good phases. The problem is to define the coordinates of the suitable centres of
these blobs. For every FAM one can calculate the corresponding structure
factors. The closeness of the calculated magnitudes to the corresponding
observed values reflects to some extent the model quality. Nevertheless, a
straightforward search for blob positions, which optimise some formal criterion
(e.g. maximise the magnitude correlation coefficient) often leads to a false
optimum and the corresponding phases do not match the true ones. As an
alternative to the global optimization, a procedure of Monte Carlo type may be
suggested [3, 4, 8], which includes the following steps:
- a large number of FAMs are generated randomly and a set of structure
factors is calculated for each generated model;
- if the correlation of the calculated and observed magnitudes is large
enough, then the phases corresponded to this FAM are announced "admissible" and
stored for further analysis;
- the selected phase sets are grouped into "clusters" of close phase sets;
- the average phase values are calculated for each of isolated clusters.
A small number of sets of the averaged phases (one set for one cluster) are
considered as alternative (for the moment) solutions of the phase problem. It
should be noted that an essential feature of cluster analysis and phase
averaging is an alignment of phase sets in accordance with the set of admissible
shifts of the origin [2,6].
Sometimes cluster analysis reveals only one significant cluster and averaging in
this cluster provides a unique (for the current step of the study) solution of
the phase problem. If several clusters have been found, the problem arises to
choose the best one. For each cluster the averaged phases (together with the
observed magnitudes) may be used to calculate Fourier synthesis and to determine
a molecule envelope on the basis of this synthesis. The likelihood based choice
may be used as a way to choose out of alternative envelopes. In brief, the idea
of the likelihood based choice may be explained as follows. Let some region in
the crystal unit cell be specified. We hope that this region is an approximate
envelope of the molecules, i.e. the majority of atoms are located in this
region. The soundness of this hypothesis may be checked with the following
statistical test. Let the atomic coordinates be chosen randomly in the tested
region and the structure factors corresponding to this random model be
calculated. The values of these structure factors are random variables and we
may inquire what the probability for the calculated magnitudes to coincide with
the observed ones is. One may believe that this probability will be high if the
tested region does contain the most part of real atom positions. On the
contrary, if the region has nothing to do with the real molecules envelope, then
the atoms placed in this region will hardly ever reproduce the observed
magnitudes. The probability for the calculated magnitudes to be equal to the
observed ones will be low in this case. The probability discussed is nothing
else than a statistical likelihood corresponding to the hypothesis that the set
of experimental magnitudes may be considered as the magnitudes calculated from
the model generated randomly inside the tested region. If several alternative
regions are suggested as candidates for a possible molecule envelope, then the
likelihood may be calculated for each of them. The choice of the region which
provides the maximal likelihood is just the realisation of the maximal
likelihood principle, which is widely used in mathematical statistics. In
practice, the value of the likelihood corresponding to a particular envelope may
be estimated by a specially designed computer simulation procedure of
Monte-Carlo type.
Statistical modelling and likelihood based methods may be used for solving of
some other crystallographic problems too.
The approach developed was used in low resolution phasing of ribosomal T50S
particle [5, 7, 10, 11, 13] and low density lipoprotein [12].
The main results concerning the development of this approach were summarised in
N.Lunina's PhD thesis [10, 11].
March, 24, 2003
V.Lunin
Publications
The full texts of papers
- Lunin, V.Yu., Urzhumtsev, A.G. & Skovoroda, T.A. (1990). "Direct
low-resolution phasing from electron-density histograms in protein
crystallography". Acta Cryst., A46, 540-544.
- Lunin, V.Yu. & Woolfson, M.M. (1993). "Mean Phase Error and the Map
Correlation Coefficient". Acta Cryst., D49, 530-533.
- Lunin, V.Yu., Lunina, N.L., Petrova, T.E., Vernoslova, E.A., Urzhumtsev,
A.G. & Podjarny, A.D. (1994). "On the ab-initio solution of the phase problem
for macromolecules at very low resolution. The Few Atoms Model method". Joint
CCP4 and ESF-EACBM Newsletter on Protein Crystallograph, 30, 37-44.
- Lunin, V.Yu., Lunina, N.L., Petrova, T.E., Vernoslova, E.A., Urzhumtsev,
A.G., Podjarny, A.D. (1995). "On the ab-initio Solution of the Phase Problem for
Macromolecules at Very Low Resolution: the Few Atoms Model Method". Acta Cryst.,
D51, 896-903.
- Volkmann, N., Schlunzen, F., Urzhumtsev, A.G., Vernoslova, E.A., Podjarny,
A.D., Roth, M., Pebay-Peyroula , E., Berkovitch-Yellin, Z., Zaytzev-Bashan, A. &
Yonath, A. (1995). "On ab-initio phasing of ribosomal particles at very low
resolution". Joint CCP4 and ESF-EACBM Newsletter on Protein Crystallography, 31,
23-32.
- Lunin, V.Yu. & Lunina, N.L. (1996). "The Map Correlation Coefficient for
Optimally Superposed Maps". Acta Cryst. A52, 365-368.
- Urzhumtsev, A.G., Vernoslova, E.A. & Podjarny, A.D. (1996). "Approaches to
Very Low Resolution Phasing of the Ribosome 50S particle from Thermus
thermophilus by the Few-Atoms-Models and Molecular-Replacement Methods". Acta
Crys., D52,1092-1097.
- Podjarny, A.D., Urzhumtsev, A.G. & Lunin, V.Y. (1997). "Model based low
resolution phasing". In: Direct Methods for Solving Macromolecular Structures,
ed. S.Fortier, NATO ASI Series C, Vol.507, 421-431.
- Lunin, V.Yu., Lunina, N.L., Petrova, T.E., Urzhumtsev A.G. & Podjarny A.D.
(1998). "On the Ab initio solution of the Phase Problem for Macromolecules at
Very Low Resolution. II. Generalized Likelihood Based Approach to Cluster
Discrimination". Acta Cryst. D54, 726-734.
- Lunina, N.L. (1998). "Computational approaches to the solution of the low
resolution phase problem in macromolecular crystallography". Resume of Ph.D.
These, ONTI PNC RAN, Pushchino, Russia. (In Russian)
- Lunina, N.L. (1998). "Computational approaches to the solution of the low
resolution phase problem in macromolecular crystallography". Ph.D. Theses, ITEB
RAS, Pushchino, Russia. (In Russian)
- Lunin, V.Y., Lunina, N.L., Ritter, S., Frey, I., Berg, A., Diderichs, K.,
Podjarny, A.D., Urzhumtsev, A. & Baumstark M.W. (2001). "Low-resolution data
analysis for low-density lipoprotein particle". Acta Cryst., D57, 108-121.
- Lunin, V.Y., Podjarny, A.D. & Urzhumtsev, A. (2001). "Low-resolution
phasing in macromolecular crystallography". In : Advances in Structure Analysis,
CSCA, Prague, Czech Republic, R.Kuzel & J.Hasek, eds., 4-36.
|
